![]()
💡 Next-generation sequencing technologies, particularly metabarcoding and metagenomics, have revolutionized microbial ecology and enhanced our understanding of the human gut microbiota. However, methodological biases in these approaches can compromise reproducibility across studies.
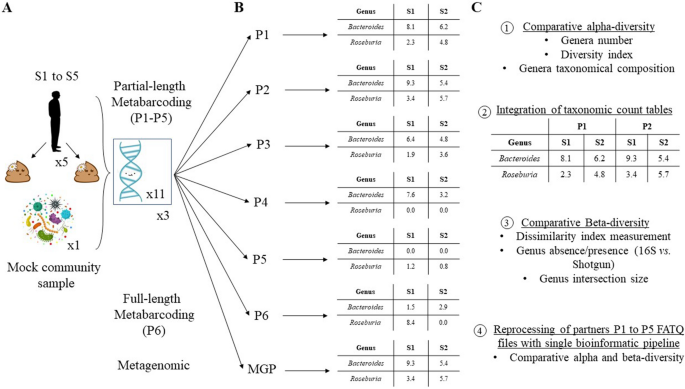
This multicenter study involved seven laboratories employing partial-length metabarcoding (P1 to P5), full-length metabarcoding (P6), or metagenomic profiling (MGP) to analyze DNA from a mock microbial community and fecal samples. They assessed inter-laboratory reproducibility, focusing on sequencing protocols and bioinformatics pipelines.
📍 Methods:
Fecal samples from five donors were collected at two time points, and DNA extraction followed the Human Microbiome Standards (IHMS) protocol. The mock and isolated DNA were sequenced by participating laboratories. Taxonomic-count tables at the genus level were analyzed, and alpha- and beta-diversity were assessed. Raw sequences from partial-length metabarcoding partners were reprocessed using a unified bioinformatics pipeline for comparison.
📍 Key Findings:
📌 Alpha-Diversity Variations: Large alpha-diversity variations among laboratories were detected, unrelated to sequencing depth. P1 identified genera unique to them, while two-thirds of MGP genera were missed by P3.
📌 Beta-Diversity: Inter-individual variance was lower than inter-laboratory variances. P5 and P6 profiles were more similar to MGP than others. Reanalysis of P1-P5 sequences using a single pipeline harmonized bacterial profiles.
📌 Bioinformatics Impact: Significant differences in alpha-diversity were attributed to bioinformatics pipelines. P3 showed discrepancies corrected by reprocessing, suggesting primer or PCR issues. Greengenes database use by P1 and P3 influenced results, highlighting the importance of database choice.
📌 Full-Length 16S rRNA Gene: Full-length metabarcoding reduced exclusive genera presence and improved similarity to MGP. However, unclassified sequences were high, possibly due to database limitations.
📌 Recommendations: Systematic in-silico testing of primer, PCR, and database combinations is advised. Standardization efforts should include reference standards, such as gut DNA mock communities, to enhance reproducibility and inter-laboratory homogeneity.
📍 This study underscores the impact of sequencing protocols and bioinformatics pipelines on gut microbiome profiling. Biases due to library preparation and taxonomic annotation databases in metabarcoding are substantial. Standardization efforts should consider both aspects, and the use of reference standards is crucial for accurate microbiota profiling.
Link to the article : https://tinyurl.com/mrbk4sfe